
How to Analyze Your Data with PAUSE3
Once you’ve run your analysis in PAUSE, you may be wondering how to interpret the plots you receive. This short guide should assist you in just that.
Good Results
After running PAUSE Plotter on raw sequencing data, good quality data will produce output that looks like the following:
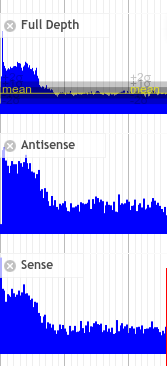
Here the “Full depth” is just the sequencing coverage of every base in the genome. It gives us a region of interest to zoom in on.
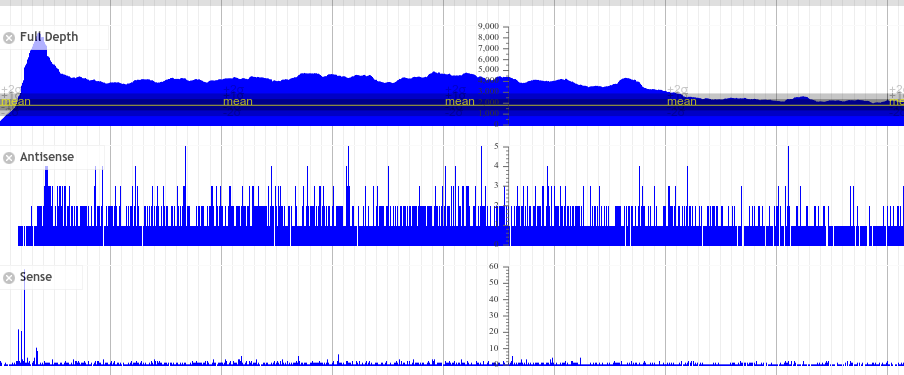
This looks somewhat like what you’re probably used to!
The “full coverage” track corresponds to both the sense + antisense mapped reads merged together, then we just display the read start tracks as the read-end tracks did not provide useful data in PAUSE2.
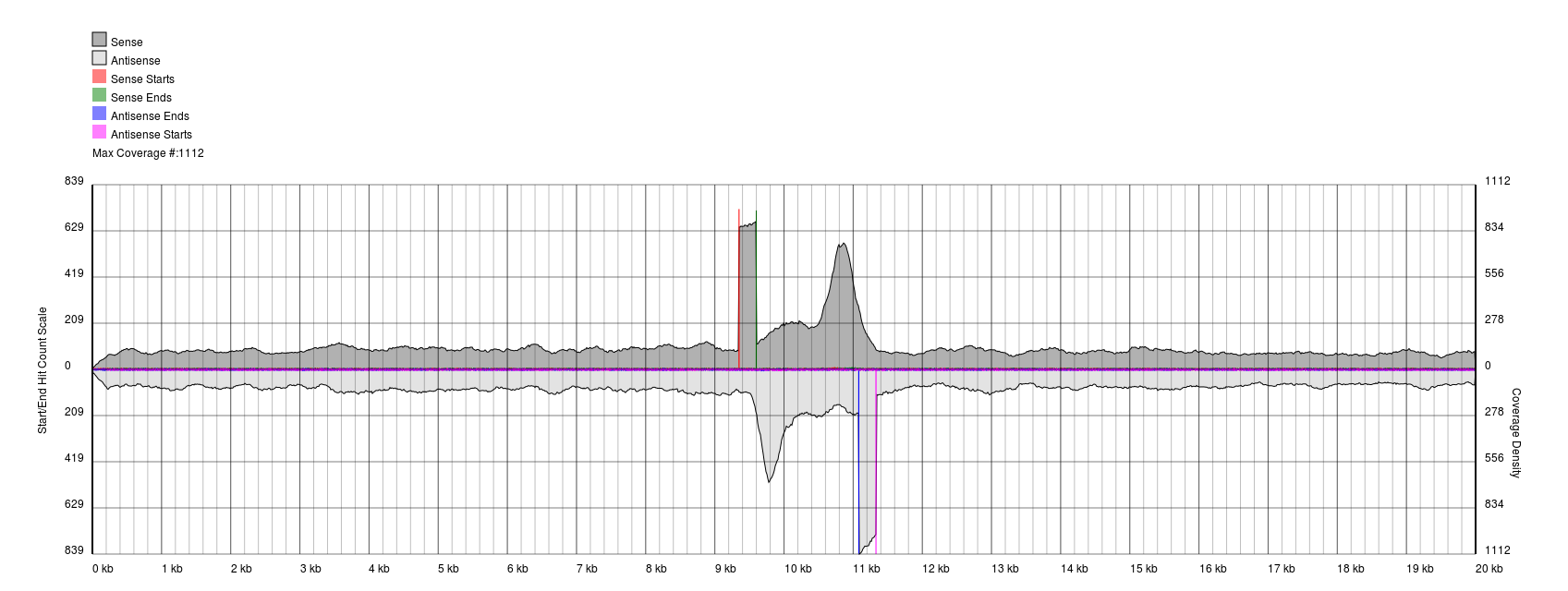
With a good quality start, we can even zoom down to the individual base level and see exactly where the reads start to pile up:
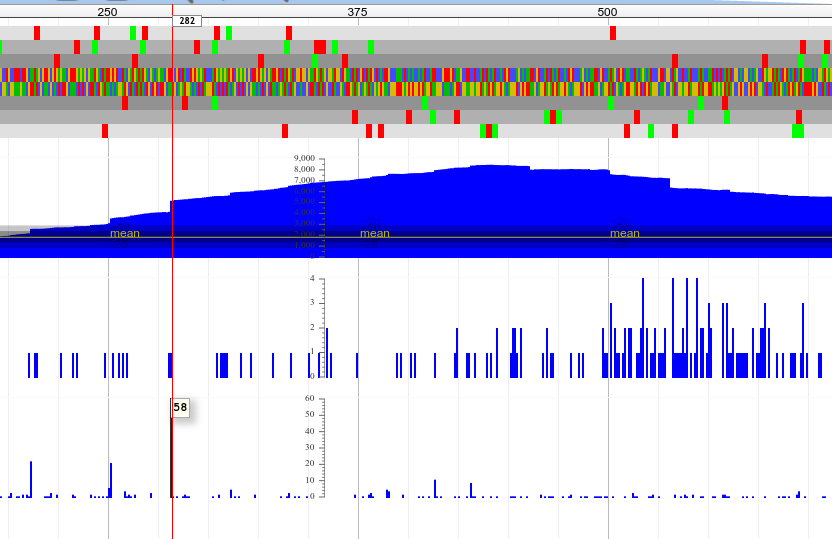
Here we can see a spike to 58 on the sense track, which corresponds to base 282 in the genome.
Bad Results
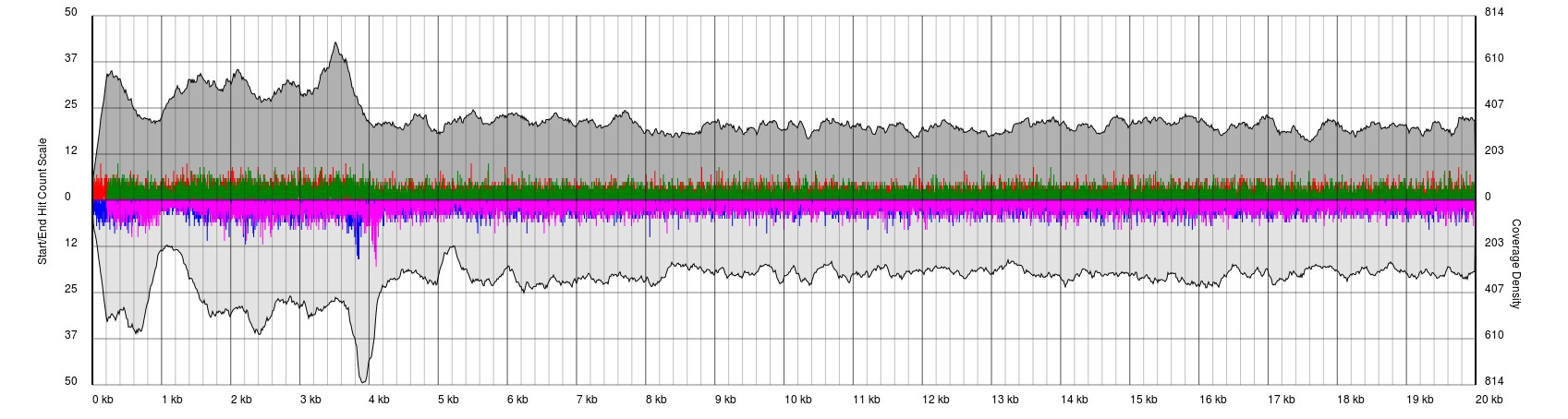
Data sets will appear noisy and you will find no defined sense or antisense starts. In truth, the read start data is no more noisy than in good quality data, however due to the much lower sequencing depth, the spikes are significantly higher relatively to the sequence coverage plots (gray). In this case it is not possible to see pile-ups
There are endless possible failure modes with no interpretation, take for example the following:
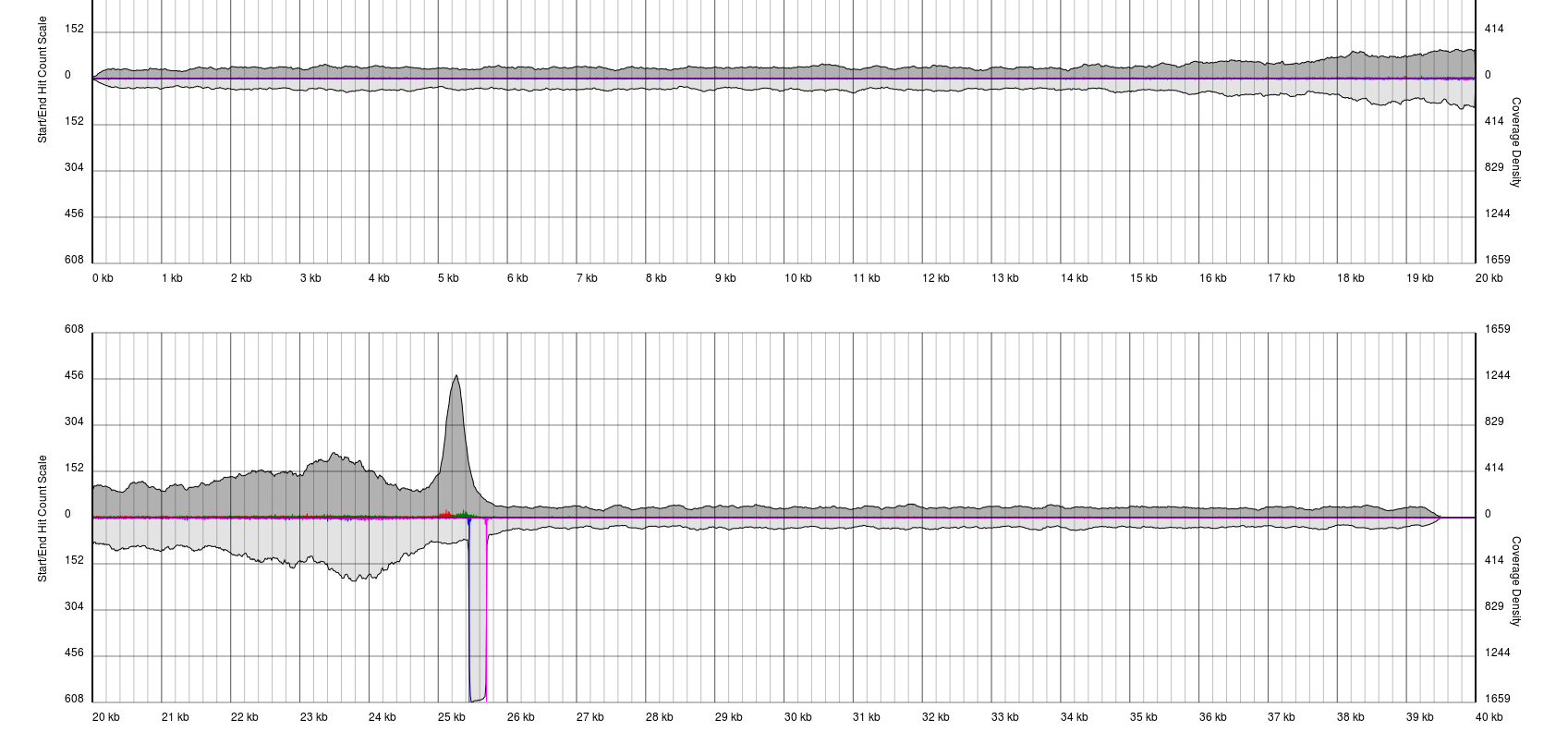
In other cases, you will see a gradual pile-up of starts or ends and will not be able to confidently predict either the sense/antisense start/end to the terminal repeat. In these cases, terminal redundancies cannot be predicted. Experimentation or re-sequencing will be needed to determine genomic ends or terminal redundancy boundaries, respectively.
Quick Guide
| Type | Description |
|---|---|
| Long TRs | Characterised by regions of double coverage >1.5kb |
| Short TRs | Characterised by regions of double coverage 0-1kb |
| Non-Cos/Headful packaging strategies | No defined appearance in PAUSE and require PCR for closure. |
Future Work
We’ll be bringing back the automated peak calling hopefully soon! Stay tuned for updates.