Annotation In Apollo
Overview
QuestionsObjectives
Requirements
Time estimation:
Agenda
- Prerequisites
- Making an Annotation
- Ensure Changes in Gene Information are Saved
- Making the Best Prediction
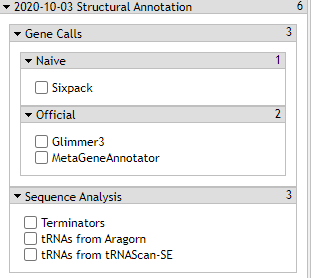
- Gene Calls
- BLAST
- NT (Nucleotide) database
- NR (non-redundant) protein database
- Phage Analyses
- Candidate ISPs/OSPs
- Possible Intron Locations
- Possible Frameshifts Sequence Analyses
- InterProScan
- TMHMM (Transmembrane hidden Markov model)
- Terminators
- tRNA and tmRNA
Prerequisites
Before beginning annotation within Galaxy (CPT Public Galaxy, CPT TAMU Galaxy), it is necessary that there is a genome loaded into Apollo. Additionally, the structural annotation must be complete, and the functional annotation workflow has already been run. The functional annotation workflow opens up the necessary evidence tracks for annotation.
Making an Annotation
Generally, the annotation process is a synthesis between the understanding of phage genomics and the available evidence tracks. The Center for Phage Technology encourages phage annotators on the CPT’s Apollo instance to follow some specific conventions (Field -> Recommended Input):
- Name -> Gene name (Could be something like terminase small subunit or hypothetical protein.) Follow the universal naming conventions at NCBI and UniProt.
- Symbol -> Do not use.
- Aliases -> Do not use.
- Description -> Do not use.
- DBXRefs -> Only use if the annotator is experienced; please ensure formatting is correct.
- Attributes -> Do not use, except in spacial cases such as for frame shifted proteins.
- PubMed IDs -> Do not use.
- Gene Ontology IDs -> Do not use.
- Comments -> Apply any free-text comments here. (Could be something like the e-value(s) between the annotated gene and homologs or notes to one’s self.)
Note that…
Calling genes is covered in another tutorial.
To annotate a gene that has been called, right click on the gene in the pale yellow User-Created Annotations track, and select “Open Annotation (alt-click).”
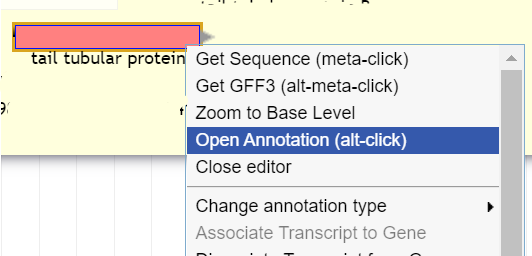
A screen will appear to the right of the main view with various fields that can be filled in with information about the gene.
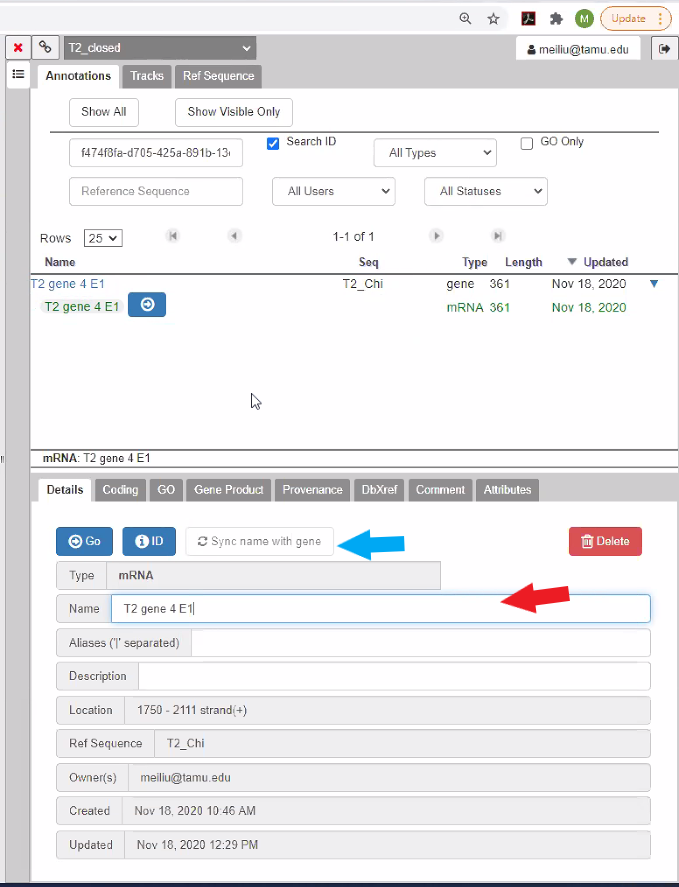
Reference the list above to see how the CPT would prefer to have genes annotated. Note that in the current Apollo verion, when you select the gene under “Name” in the annotation screen shown above, the gene name is NOT directly editable. Instead, only the mRNA name is editable and the annotated mRNA name is what is displayed in the annotation track.
Click on the mRNA and in the mRNA editing window (bottom portion of the annotation screen), enter your chosen name for the mRNA in the “Name” box (red arrow), and hit ‘enter’. You may see a Save password dialog box, if you do just hit ’No thanks’ or close the box. You should then see that the name of your mRNA in the main viewing pane has been updated. Click on “Sync name with gene” (blue arrow) in the annotation pane to make sure both the gene and the mRNA are identically named. You need to refresh the annotation tab (by clicking on a different tab in the annotation pane) in order to see the gene name syncing with the mRNA. If you experience any problems with the names not syncing in the main view or the annotation pane, then refresh the page. We are working on improving syncing the gene and mRNA name for better user experience. If you failed to sync all mRNA with gene names or choose not to sync while you annotate in Apollo, we do have a Promote Qualifier tool (TAMU user tool link) to sync all mRNA names with gene names in the Gff3 file retreived from Apollo record for downstream analysis.
Naming Guidelines
It is imperative to follow suit with the UniProt and NCBI international naming conventions. It allows for standardization and consistency in naming proteins, subsequently aiding data retrieval and improving communication. Follow the convention for capitalization and hypothetical protein naming.
Ensure Changes in Gene Information are Saved
There are occasional small bumps on the road when annotating in Apollo, many of which are encountered when editing information for a gene. It helps to be aware of how to avoid them, and where to fix issues when they arise. When changing a data field in the annotation, make sure that the data is saved before moving on. By clicking another field or hitting the tab key, changes should automatically save. If it has successfully been saved, the change will be immediately noticeable in the User-Created Annotations track, even without closing the annotation window. If the changes are not being saved, refresh the page and try again. If this continues to happen, try opening Galaxy (CPT Public Galaxy, CPT TAMU Galaxy) in an incognito window.
Making the Best Prediction
The Center for Phage Technology integrates as many data sources as is feasible when making predictions about gene function. Please contact the CPT IT staff (cpt@tamu.edu) if another data source not currently available in Apollo is desired. The CPT staff will assess your recommendation and may add it to the Phage Annotation Pipeline (PAP).
A word on genome annotation
While we use all the best bioinformatic tools available to complete these analyses, all these are still predictions. Hypothetical. Every prediction is subject to being wrong and getting corrected with new information. This is the nature of genome annotation, and science in general. Due diligence and thorough work is expected, but it is inadvisable to agonize over any single gene prediction.
Many of the protein prediction tracks yield multiple homologs for the same gene. To learn more about each homolog, hover over the homolog to be investigated, or right-click on it and select “View details.”

The details presented will vary between evidence types (e.g.BLAST vs. InterProScan).
Gene Calls
The CPT’s PAP integrates gene calls from numerous sources, specifically MetaGeneAnnotator and Glimmer3. These gene callers are generally very accurate. However, should those fail to find a gene, SixPack is a backup gene caller.
Note that…
Gene calling is part of structurally annotating a genome in Apollo. For more information on structural annotation and gene calling, please look at this tutorial.
BLAST
BLAST is an acronym standing for Basic Local Alignment Search Tool. Different variations of BLAST exist to search different kinds of sequences. BLAST breaks down the input sequence (called the query) into k-mers and compares these to sequences in the database; default k-mers are currently 6 for BLASTp (protein database) and 28 for BLASTn (nucleotide sequence database). Matching k-mers are then extended to the left and right until the score drops below a threshold, T. Sometimes the extended coverage encompasses the entire sequence, and other times the alignment is broken up into homologous segments that are separated by low similarity regions. Alignments with high similarity are referred to as a high-scoring pair (HSP); this may be the entire sequence pair, or only part of the query.
Although BLAST is accessible through the NCBI website, the databases detailed below are those that are available to an annotator in Apollo. In Apollo, right-clicking on a hit and selecting ‘View Details’ will yield a summary of the details about the protein.

- Score = E-value; the lower the score, the better the alignment with the query. This is also reflected in the color highlight intensity of the feature displayed in the evidence track.
- Description = may be informative, but this depends on the quality of the annotation in the protein record. In the above example, the accession number is underlined; this same information can be found in the Description portion. Searching the accession number in NCBI BLAST will yield much more information about the protein, including the paper in which this protein was originally reported.
Note that…
In practice, an E-value of less than 1e-3 or 1e-5 are considered relevant, if that hit covers most or all of the protein!
1. Nucleotide (NT) database
Megablast is run against a copy of NCBI’s NT database. Hovering over a hit segment will show where in the target genome the region aligns. Look into the functional flow used for the exact setting (such as the Dice value cutoff when displaying the results) to better interpret the results.

2. Protein database
BLASTp is run against three databases (in the most recent PAP iteration, after PAP Functional Workflow v2020.07):
- CPT’s Canonical Phage database, a select collection of high-quality and well-studied representative phage proteomes
- SwissProt (curated from UniProt)
- nr (from NCBI). Note that the CPT use a nr database that only include viruses that infect baceria in the most recent functional workflow.
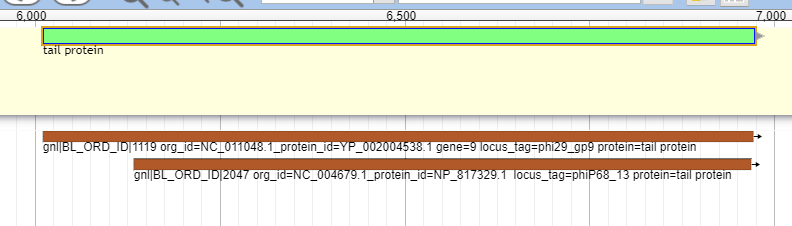
These databases offer insight into possible names and functionalities for the phage proteins being analyzed. An example of the Canonical phage track can be seen below.
Phage Analyses
The CPT has developed a number of phage analysis tools for aid in phage specific annotation. These are supplementary bits of information which can inform the analysis, but they must be looked at critically. Many of these tools intentionally yield many possible options, unfortunately yielding high false positive rates.

1. Candidate ISPs/OSPs
Phage lysis genes are notoriously poorly annotated. Often they are missed or completely misattributed. To combat this problem for phage spanin proteins, lysis proteins specific to disrupting the outer membrane of gram-negative bacterial hosts, the CPT utilizes the candidate ISP (i-spanin) and OSP (o-spanin) tool output.
Note that…
These tracks will generate false positives. Be sure that the data occurs somewhere around the phage’s lysis cluster (where applicable). Additionally, know what to look for in a lipobox in these potential spanin genes.
The ISP track naïvely searches the genome for every possible CDS, and then analyzes them with TMHMM. This happens even in the case of a mis-called or entirely missed i-spanin. The OSP track searches through every possible CDS which contains a lipobox as defined by the CPT. Both of these datasets are filtered for proximity. Co-incidence of a possible ISP gene and a possible OSP gene is a good sign, but the genomic context information will need to be taken into account to complete the functionality inference.
2. Possible Intron Locations
This track analyzes BLASTp against NR data for locations where two or more called, disjointed CDSs match separate locations on the same target protein. Below is an example alignment from phage K.
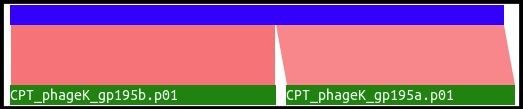
Both 195a and 195b align to distinct regions of the same protein, based on BLAST data. It can be theorized that these are actually one protein with one intron and two exons; however, this evidence should not be taken as 100% correct. Similar results may happen for other reasons, such as separation of domains from a single protein due to evolution, sequencing errors, and a myriad of other possibilities. See this tutorial on how to use the possible intron locations track to annotate interrupted genes.
Note that…
Currenty the CPT does not have a tool that can automatically detect frameshifts. Refer to this frameshifts annotation tutorial for more information on how to annotate tape measure protein chaperone frameshifts.
Sequence Analyses
Additional analyses run in the PAP are listed in the Annotations Track on the left under both the structural and functional sections.
1. InterProScan
InterProScan is an extremely useful domain finder. It is hosted by EMBL-EBI (European Molecular Biology Laboratory - European Bioinformatics Institute) and integrated into UniProt (a freely accessible database of protein sequence and functional information, of which many entries are derived from genome sequencing projects). InterProScan searches a protein sequence against the member databases and detects similarity to conserved domains. It integrates 14 other conserved domain databases to assign a single InterPro ID to related domains. Hits from InterProScan predict protein function based on conserved domains (beware of domain swapping!)
Domain Swapping
- It is not uncommon for proteins to contain more than one domain that function independent of each other. These domains can be found alone. Sometimes you may see a BLAST hit to a protein that has two domains, but the name of the protein only reflects one domain’s function. When you see a hit, make sure that the domain the name is based off is actually aligning to your query.
These InterPro conserved domain hits will be more sparse than BLAST hits and often only align to a portion of the protein. Right clicking on a feature from the InterProScan evidence track and selecting ‘View Details’ will yield a summary of that feature.

If the domain hit is part of InterPro, there will be a “Dbxref” entry. Searching either the name or Dbxref fields in Google (or use the IPR number at InterPro’s website) will often find the domain entry in either the member database or InterPro. The Score for these conserved domain searches is a probability or a quality score, and its calculation varies between databases. This tool, among all other tools food in Apollo, have been pre-calibrated to show results the authors think are significant, so the annotator does not need to evaluate this value.
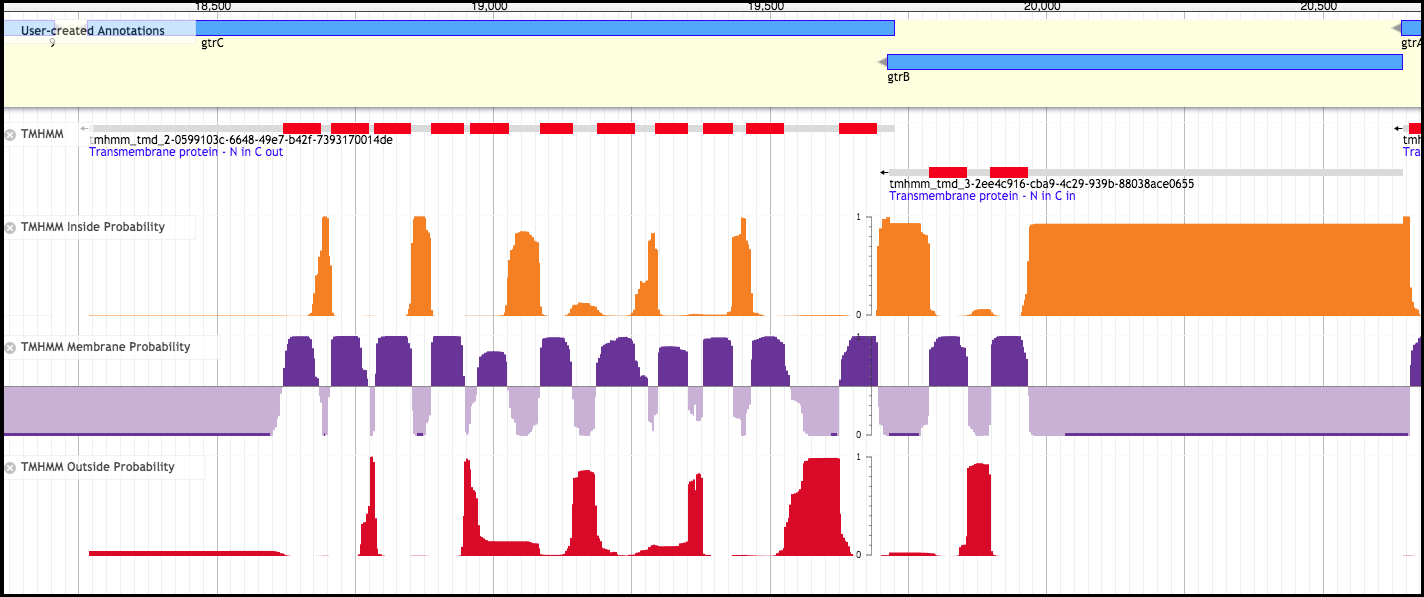
2. TMHMM (Transmembrane hidden Markov model)
Here, TMHMM is run over the genome to pick out genes that contain likely transmembrane domains (TMDs). TMHMM data is used in a number of other tracks and analyses as well.
There are three variations of the TMHMM track - Inside Probability, Membrane Probability, and Outside Probability. The Membrane Probability track yields the actual scores from the TMHMM track plotted to the genome. The Inside Probability track shows the probability of a region of a protein being within the cytoplasm. The Outside Probability show the probability of a region of a protein being within the periplasm or extracellular.
3. Terminators
Terminators are produced from TransTermHP. TransTermHP finds rho-independent transcription terminators in bacterial genomes. Each terminator found by the program is assigned a confidence value that estimates its probability of being a true terminator.
Note that…
This track can be found underneath the “Sequence Analysis” section of the Structural Annotation portion all of the tracks.
4. tRNA
ARAGORN and tRNAscan-SE provides the CPT with quality tRNA annotations. Recall that tRNAs are not likely to be embedded within genes.
Note that…
tRNA tracks can be found underneath the “Sequence Analysis” section of the Structural Annotation portion.
Congratulations on successfully completing this tutorial!
Help us improve this content!
Please take a moment to fill in the Galaxy Training Network Feedback Form. Your feedback helps us improve this tutorial and will be considered in future revisions.