
In order to help people correct and finish their genomes, we’ve built a small tool to help edit genomes. It allows you add, remove, and re-arrange sequence. Once you’ve built up your new patchwork genome, the tool outputs the re-arranged genome and re-arranged features for you.
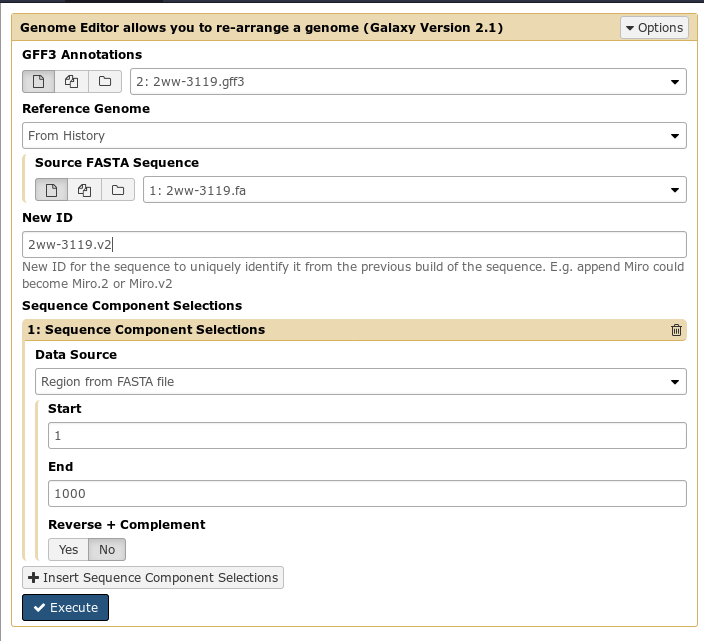
First, you select an input fasta and gff3 file for your current build of the genome. Then, you specify Sequence Component Selections (regions of a genome, composed of start, end, and whether or not it should be reverse complemented) that you wish to be in the final output file.
By doing this, you can build up a complete output genome from components of the input genome. For example, you might “select” regions 1-10000(+) and 10000-20000(-). The first region you select will come first in the output genome, the second region will be reversed and appended to the first.
Additionally, if you need to add sequence that was not included in the original genome build, you can do so easily. These can be intermixed anywhere in your new genome build creation process:

Motivating Example
As a motivating example, we provide the following situation that we encountered. A student’s genome needed to be edited to insert a sequence in the middle of the genome, and a small bit at the tail end of the genome had been mis-assembled and was inverted. We can make these modifications in a fairly straightforward manner:
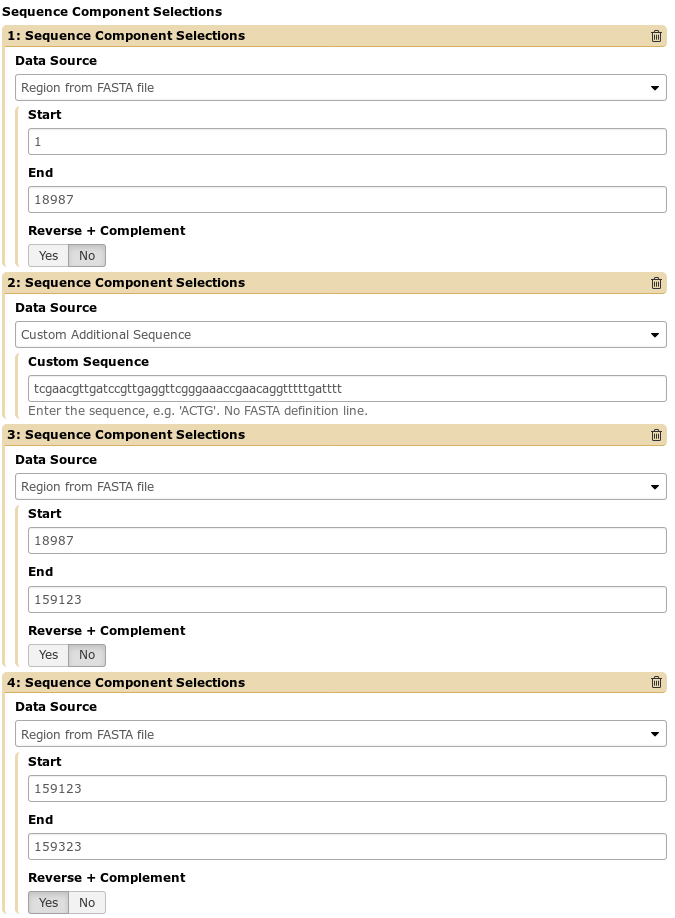
This will re-arrange the sequence and features and generate our output file exactly as we expect.

One of the outputs is a simple table indicating the regions of the input sequence that map to regions of the output sequence. For the above example, we will see the following table:
| Input Genome | Start | End | Strand | Output Genome | Start | End | Strand |
|---|---|---|---|---|---|---|---|
| 2ww-3119 | 0 | 18987 | + | 2ww-3119.v2 | 0 | 18987 | + |
| 2ww-3119 | 18986 | 159123 | + | 2ww-3119.v2 | 19037 | 159174 | + |
| 2ww-3119 | 159122 | 159323 | – | 2ww-3119.v2 | 159174 | 159375 | + |
We imagine that this could be used like a chain file to map other genomic features that weren’t included in the input gff3 file (which are re-mapped by default and available in your outputs).