3. Annotation¶
3.1. Process¶
The process for annotation generally involves synthesizing your knowledge of phage genomics with the evidence tracks available to you.
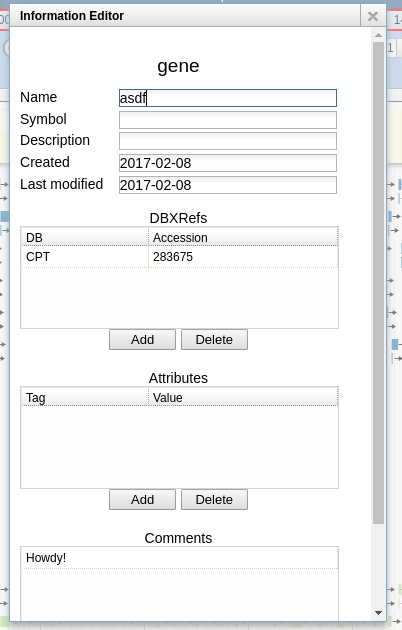
Fig. 3.5 While we have blurred out the mRNA panel, many users find it helpful to copy the name they apply to the gene feature straight to the mRNA feature. This makes the name visible when scrolling across the genome.
The CPT encourages phage annotators on the CPT’s Apollo instance to follow these conventions:
| Field | Recommended Value |
|---|---|
| Name | Your gene’s name. Could be something like DHFR or Hypothetical Novel. |
| Symbol | Do not use. |
| Description | Do not use. |
| DBXRefs | Use only if experienced annotator, please ensure format is correct. |
| Attributes | Do not use. |
| Pubmed IDs | Do not use. |
| Gene Ontology IDs | Do not use. |
| Comments | Apply any free-text comments you would like here. |
3.2. Data Sources¶
We integrate as many data sources as is faesible. If you need another data source that does not appear in Apollo, please contact Eric Rasche and the CPT can work on adding that to the Phage Annotation Pipeline (PAP).
3.2.1. Blast / NT¶
We run megablast against a copy of NCBI’s NT database.
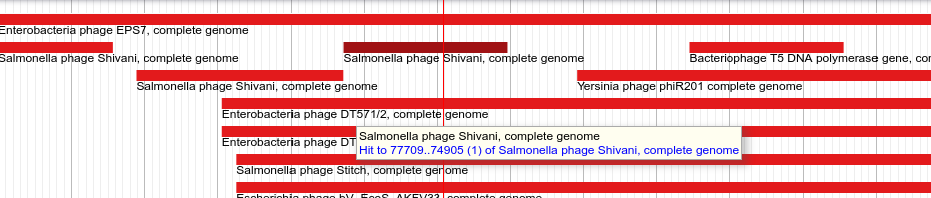
Fig. 3.6 Hovering over a hit segment will let you know where in the target genome the region aligns.
3.2.2. Blast / NR¶
We run BlastP against three databases:
- CPT’s Canonical Phage Database
- UniRef90
- nr
These databases can give you good insights into possible names and functionalities for proteins.

Fig. 3.7 The alignments are gapped, and the gaps are joined with a glyph tradationally used by intron/exon junctions. This is a known bug. Additionally there are some other display issues which are also known bugs and will be resolved in a future update.
3.2.3. Gene Calls¶
The CPT’s PAP integrates gene calls from numerous sources, specifically GeneMarkS, MetaGeneAnnotator, and Glimmer3
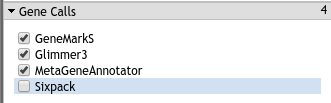
Fig. 3.8 The real gene callers are generally very accurate. Should they completely fail to find a gene you can rely on sixpack’s gene calls.
3.2.4. Phage Analyses¶
We run a number of “phage analyses” which are mostly tools the CPT has developed to aid in phage specific annotation.
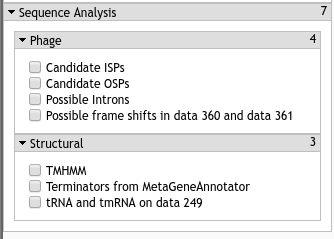
Fig. 3.9 Our phage analysis tools cover many different types of analyses. These are supplementary bits of information which can inform and analysis, but must be looked at critically. Many of our tools intentionally return many possible options, leading to high false positive rates.
3.2.4.1. Candidate ISPs / OSPs¶
Candidate ISP and OSPs are our attempts to determine possible phage spanin component locations. This track will feature a huge number of false positives, so you should be sure that the data occurs somewhere around your lysis cluster (where appropriate).
The ISP track naively searches the genome for every possible CDS, and then analyses them with TMHMM. We do this in case you miscalled, or missed your I-Spanin. The OSP track searches through every possible CDS which contains a lipobox, defined by the CPT’s regular expression for them.
Both of these datasets are filtered for proximity. Co-incidence of a possible ISP gene and a possible OSP gene is a good sign, but you will need to use genomic context information to complete the functionality inference.
3.2.4.2. Possible Introns¶
This track analyses your BlastP against NR data for locations where two (or more) disjoint, called, CDSs match separate locations on the same target protein. An example from Phage K is illuminating here:
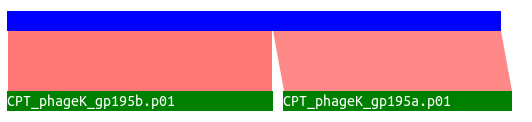
Both 195a and 195b align to distinct regions of the same protein, based on BLAST data. It can be theorized that these are actually one protein with one intron and two exons, however this evidence should not be taken as 100% correct. Similar results may happen for other reasons such as separation of domains from a single protein due to evolution, sequencing errors, and a host of other possibilities.
3.2.4.3. Possible Frameshifts¶
This track, likewise, is very optimistic in what it calls a possible frameshift. It searches for the XXXYYYZ pattern (allowing for some wobble) wherein a frameshift would not change both codons. This is based on evidence in 10.1016/j.molcel.2004.09.006.
3.2.5. Sequence Analyses¶
These analyses are other sequence or structural predictions.
3.2.5.1. TMHMM¶
Here TMHMM is run over your genome to pick out genes containing likely TMDs. TMHMM data is used in a number of other tracks and analyses as well.
3.2.5.2. Terminators¶
Terminators are produced from TransTermHP. You cannot currently annotate these in Apollo. That is being worked on upstream. For now, when you go to publish a genome, the CPT will work with you to run a number of other automated annotation processes as part of a post-processing step.
3.2.5.3. tRNA and tmRNA¶
ARAGORN provides us with quality tRNA annotations. You should feel comfortable annotating those in Apollo. Bear in mind that tRNA are not likely to be embedded within genes.
3.2.5.4. InterProScan¶
Missing from this graphic is InterProScan which is an extremely useful domain finder. The results from InterProScan can help inform protein identies and provide solid evidence for functionality.
InterProScan produces IPR##### entries which will be automatically applied to your genome as part of a finishing process. Please contact Eric for more information.
