Class 2 Slides: Functional Annotation¶
Introduction¶
Introduction to Functional Annotation I¶
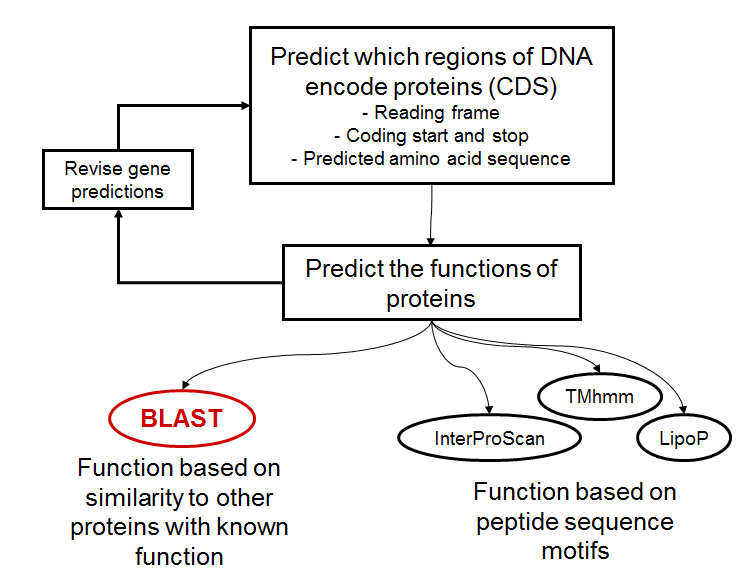
Annotation overview
Introduction to Functional Annotation II¶
- After predicting where your genes are and their product’s primary structure, we need to predict what they do
- Only possible once structural annotation is done
- The quality of your functional annotation is highly dependent on the quality of your structural annotation!
- If you are missing a gene it will not be annotated at all
- If the gene start is incorrect, this may affect the quality of an annotation but is usually fixable later
- These analyses are based on protein sequence data
- No longer creating gene models, now adding annotations to existing gene models
Why do we do Functional Annotation?¶
- A sequence alone isn’t informative, and we can’t do experiments on each gene in your genome
- However, we can often predict (guess) the function of a protein based on its similarity to other proteins that have experimental evidence
How?¶
Major methods at our disposal¶
- Directly compare the sequence of your protein to a database of previously identified proteins
- Basic local alignment search tool (BLAST)
- Compare the sequence of your protein to a model of other proteins of known function
- InterProScan
BLAST¶
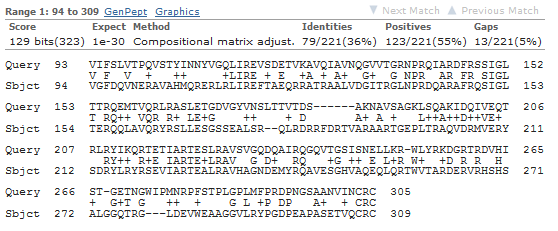
BLAST result: a single pairwise alignment of your sequence (query) and a protein in the nr database (subject)
BLAST: Limitations¶
- With BLAST, we can see that two proteins are related
- For functional annotation with BLAST, there are two major failure modes:
- The match is only partial and does not encompass that part of the protein that has function (domain swapping)
- The match is good and complete, but the target protein was annotated incorrectly (garbage in, garbage out)
BLAST: Domain swapping¶
Many proteins are organized into functional domains, each of which contributes to the protein’s function. Domains can be reshuffled to form proteins with new functions..
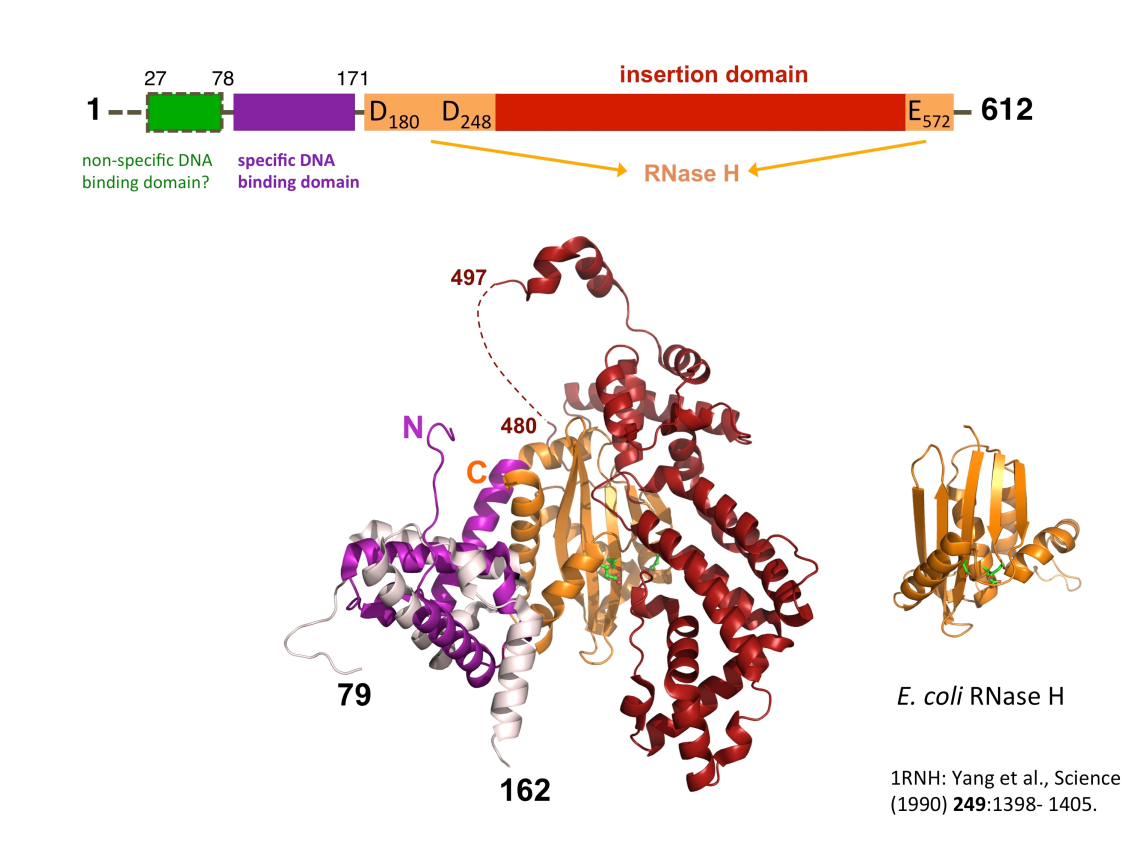
Domains in E. coli RNase H
BLAST: Domain swapping II¶
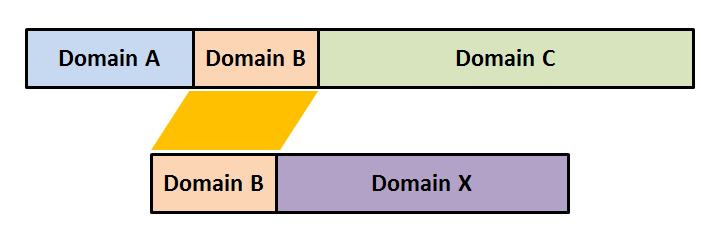
Partial protein similarity can lead to misleading results. BLAST similarity or E-value can be misleading if there is a good match over part of a protein.
BLAST: Database choice¶
- The default BLAST database, nr, is full of poorly annotated proteins
- Public repository, little quality control
- We can solve this issue by using BLAST to search against databases that only contain only proteins with high quality annotations (curated databases)
- Databases with evidence are reliable and trustworthy; other databases should be taken with a grain of salt
- Our trustworthy databases: Uniprot, custom curated phage database
InterProScan¶
About InterProScan¶
- Collection of protein models from “member databases” such as PANTHER.
- Models usually describe protein domains, domains imply functionality
- InterProScan is a wrapper that searches your proteins against all of these models and returns homogeneous results (very important in bioinformatics)
- Maintains a large database of essentially connections between models,
referenced as “IPR Numbers”,
IPR:######
Domain searches¶
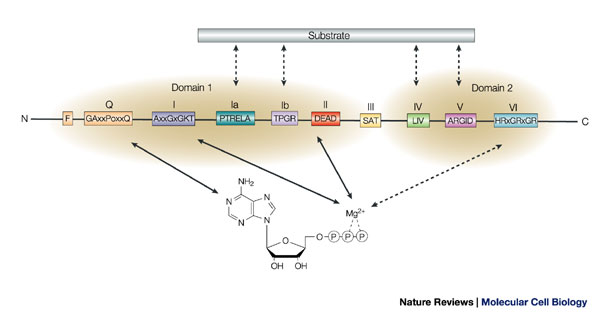
DEAD box helicases are very diverse but all have certain features in common. These features are too small to be detected by BLAST but can be detected by other tools like InterProScan.
InterProScan Results¶
- The results of an InterProScan search will appear as a track in Apollo, with dozens or hundreds of features.
- Each of these hits will have an IPR Number, a database cross-reference (dbxref) to the InterProScan database.
- We will provide you with a tool which will automatically add these to your proteins
- You will use the InterPro domains and other results to inform your protein name, and when applying GO annotations.
Blast¶
About BLAST¶
- Basic Local Alignment Search Tool
- Compares nucleotide or protein sequences to databases and calculates probability of seeing that sequences.
Scores and E-Values¶
- Quality of alignment is represented by the score
- Significance of an alignment is described by the e-value
- We focus on the e-value. This represents the number of different alignments with equal or better scores, that would occur in an identical database search by chance.
- Note the phrasing, the same query on a different database will have a different e-value.
- E-values are not P-values
BLAST Hits in Apollo¶
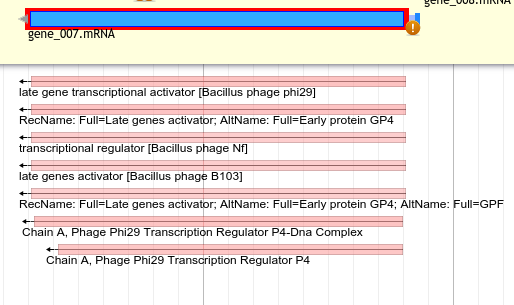
BLAST against NR
Inspecting a BLAST result¶

Inspecting an individual blast result. All identical proteins in NR are squashed together in a blast result, here we can see the other protein names
BLAST Result Tracks:¶
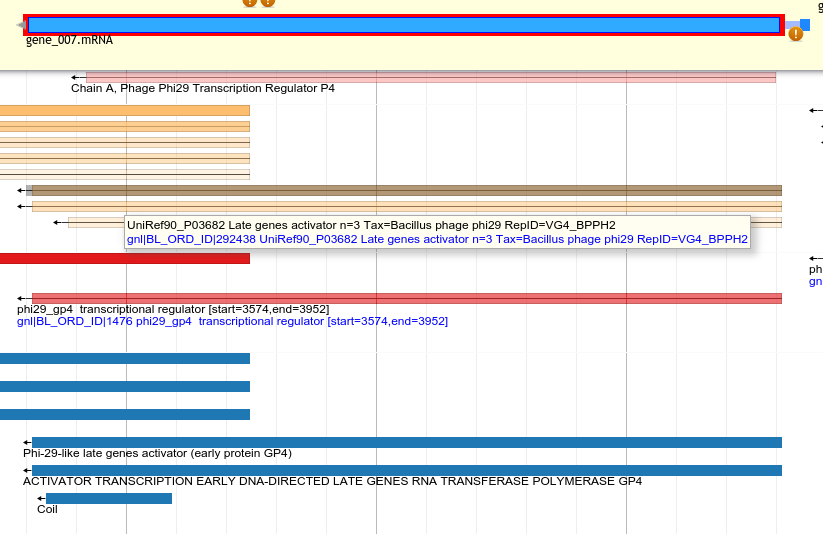
Uniref90 in yellow, canonical phage DB in red, InterProScan in blue
Annotating¶
Prerequisites¶
- Should have started workflow A by now
- Time at end of class to help get everyone up to speed
Today’s Plan¶
- This Lecture
- Quick Blast Exercise
- Annotation Free Time
Blast Exercise¶
- Available on the web, and in your Google drive and on the web.
Annotation Free Time¶
- Finish workflow A if you have not
- Ask questions!
- A more full fledged annotation guide will be posted in the Google drive and an email sent out.
- After you’ve read the annotation guide, start annotating proteins in your genome.
