Class 1 Slides: Structural Annotation¶
Annotation¶
What is annotation?¶
- Annotation is the interpretation of DNA sequence to predict its biological function
- Annotation is typically divided into two phases:
- Structural (where are the genes?)
- Functional (what do the genes do?)
- All of the annotations you make in the class will be predictions only (not based on experimental evidence)
Basic annotation overview¶
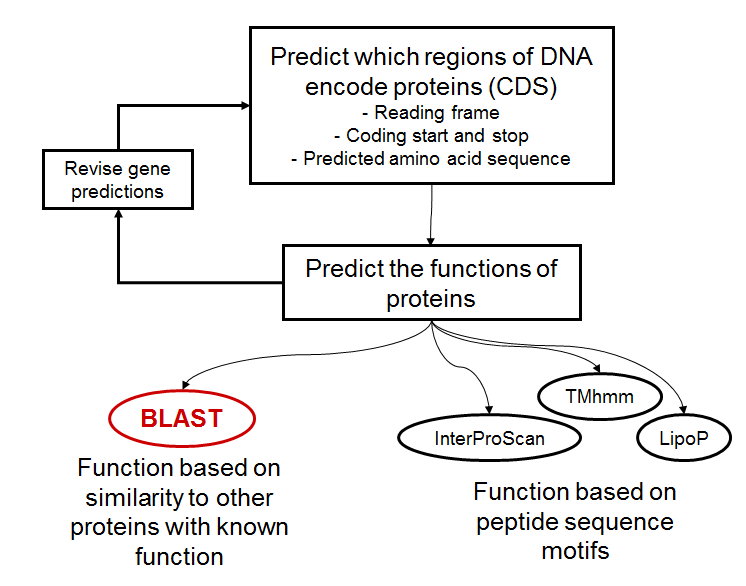
Model annotation
Objectives¶
Today¶
- Genes
- What is a gene?
- What does a gene look like?
- How do we find genes in DNA sequences?
- Manual annotation of small genomic region in-class
- Ready to annotate your own phage genome
Genes¶
What is a Gene?¶
- “A heritable unit that can be transferred from parent to offspring”
- A very eukaryo-centric view!
- What does this mean at the DNA level?
- In eukaryotic genomes, the protein-encoding “gene” is usually defined as the entire mRNA transcript and its regulator, and contains the many introns and exons that encode a protein
- Inference of the final protein sequence is difficult without additional evidence
- “Gene” does not equal “coding sequence”
What is a gene?¶
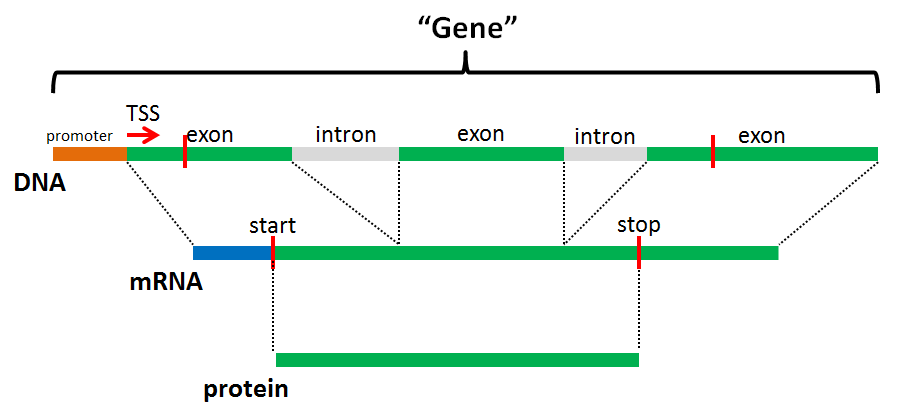
Model eukaryotic gene
What is a Gene?¶
- In prokaryotes and phages, introns are rare, and most mRNA transcripts are polycistronic
- Easy to infer protein sequence directly from DNA sequence
- The protein-encoding gene is usually depicted as equivalent to the coding sequence (CDS)
- Note that not all genes are protein-encoding!
- tRNAs, rRNAs, etc. are also encoded by genes, but in these cases the RNA is the functional product
What is a Gene?¶
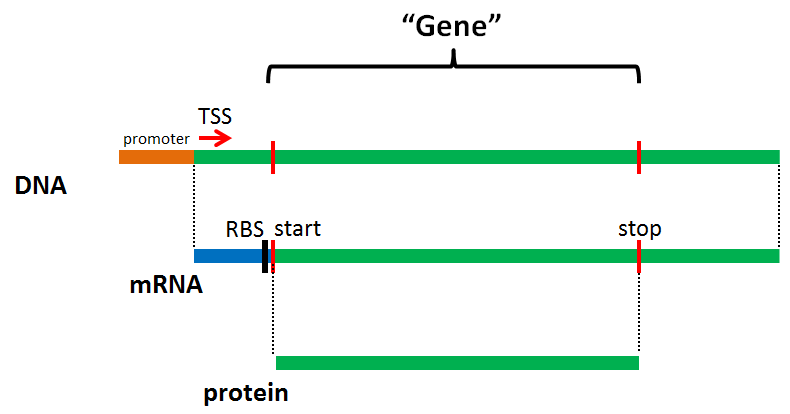
Model prokaryotic gene
What does a gene look like?¶
- The first step of our annotation process will look only for protein-encoding genes (CDS)
- A CDS must (usually):
- Have a translational start signal upstream of a valid start codon
- Encode a protein in the reading frame determined by the start codon
- Be terminated by a stop codon
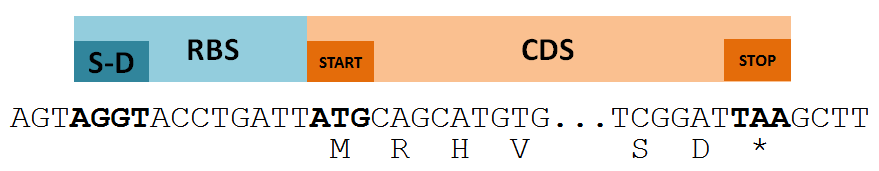
model gene DNA level
How do we find genes?¶
- If we look at all possible ORFs in a DNA sequence, many ORFs can
satisfy these criteria
- Obviously only a few can be “real” CDSs
- This kind of six-frame translation, or naive gene calling, is not adequate
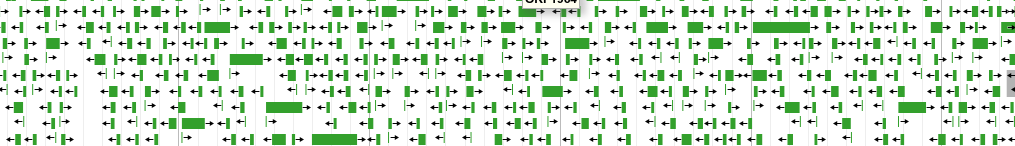
All possible CDSs in a region, many are wrong!
How do we find genes?¶
- If we filter the previous output and only keep ORFs with an RBS, there are still many possible CDSs

Filter the previous listing by presence of RBS
How do we find genes?¶
- In practice, we use automated gene calling tools that process the DNA
sequence and predict the locations of protein-encoding genes
- GeneMark, Glimmer3 and MetaGeneAnnotator are the three tools we use
- Note that these tools are much more sophisticated than simply looking
for valid gene starts
- Codon usage, dinucleotide frequencies, and conformation to models of known genes are used to predict CDSs
- These tools are relatively accurate but can still make mistakes, so your expertise is needed to correct them!
MGA Predicted Genes¶
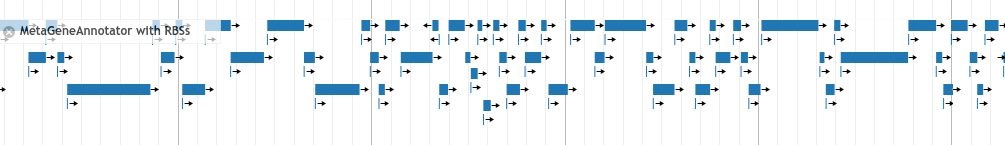
MGA predicted gene starts
What do “real genes” look like?¶
- Most genes will have a detectable RBS with a S-D sequence
- Most genes will use an ATG, GTG or TTG start
- Phages often use polycistronic mRNA’s, so genes are often clustered together on the same strand
- Phages usually want to maximize coding density, so CDSs often touch
or slightly overlap
- Don’t panic if you can’t find a CDS to fill every bit of DNA, these may contain tRNAs or regulatory elements
A note on codons¶
- Important differences in how organisms translate codons to amino acids–codified into “translation tables”
- For structural annotation, we are most interested in how the phages use start codons
- You are probably familiar with the “standard” translation
table
- Three starts (ATG, CTG, TTG) and three stop codons (TAA, TAG, TGA)
- We use the “The Bacterial, Archaeal and Plant Plastid
Code”
table
- Technically, seven starts (ATG, GTG, CTG, TTG, ATT, ATC, ATA) and three stops (TAA, TAG, TGA) are recognized
- In practice, only ATG, GTG and TTG are known to occur with any frequency.
Start Codons¶
Start codon frequencies in E. coli K-12 [BPB+97]
Codon Percent ATG 83% GTG 14% TTG 3% 1 ATT start was identified
Shine-Dalgarno Sequences¶
- An RBS is an important part of locating correct gene starts
- Valid RBS: any subsequence 3 bp or greater of the consensus S-D
sequence
AGGAGGT, spaced 4-12 bp upsteam of a start codonAGGA,AGG,GGA,GAGGare all valid
- Remember that wobble base rules apply (G-U base pairing), so
GGG,GGGG,AGGGGG, etc. are also valid
ShineFind¶
- CPT program run automatically as part of workflow
- Annotates possible S-D sequences for a set of genes
- Will help you identify a good gene call from a bad one
- Be very suspicious of gene starts with no RBS!
Exercise¶
Annotating a Real Sequence¶
- You’ll now annotate a real sequence
- Use the information you’ve learned to inform your decisions
Running The Exercise¶
- You must have completed C0 first.
- Please refer to the other handout you received, C1-exercise.
- Raise your hand if you have questions!
- Also available at
https://cpt.tamu.edu/bich464/doc/C1-exercise.html
Your Phage¶
Meet your phage!¶
- In the shared Google Drive Folder, you will find a copy of your Phage’s Genome, under the folder “Student Genomes/Your-Initials/”
- You will need to download it from Google Drive, and then upload it to Galaxy
Next Steps: Pre-requisites¶
- You will need to launch workflows in the next steps.
- Please ensure that you know how to do that.
Next Steps: Structural Annotation¶
- Import the BICH464 PAP 2016 Workflows
- Run the BICH464 PAP 2016 Part A workflow on your own genome
- Once that’s done, you will need to make your gene calls.
- Gene callers will help significantly, but remember, they’re not always right!
- Use what you’ve learned and practiced in class.
Next Steps: Functional Annotation Workflows¶
- When you’re happy with your gene calls, you’ll start workflow Part
B that you’ve imported.
- It automatically renumbers your genes
- And loads your annotations into Galaxy
- After Part B finishes (Should only take a minute or two), you’ll
start Part C
- This will take a long time, you should automatically receive an email when it is done.
References¶
| [BPB+97] | F. R. Blattner, G. Plunkett, C. A. Bloch, N. T. Perna, V. Burland, M. Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor, N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y. Shao. The complete genome sequence of Escherichia coli K-12. Science, 277(5331):1453–1462, Sep 1997. |
